Nous sommes actuellement environ 7 milliards d’humains sur Terre, appartenant tous à la même espèce (Homo sapiens). L’étude de notre génome et la comparaison de certaines de nos caractéristiques génétiques nous apporte de nombreuses informations et nous permet notamment de retracer notre histoire, à l’échelle individuelle et collective.
| La découverte de l’ADN Voici une vidéo présentant l’origine des gènes présents dans l’ADN |
Connaitre notre génome : le séquençage du génome humain
Séquencer un génome consiste à connaitre l’enchaînement des nucléotides de l’ensemble de l’ADN (la séquence de nucléotide de tous nos chromosomes). Après 15 ans de travail, le projet « génome humain » parvient à séquencer le 1er génome humain en 2004.

Le génome d’un homme moderne (Homo sapiens) comporte 3 milliards de paires de bases, réparties sur 46 chromosomes (23 paires homologues). Nous possédons environ 20 000 gènes dont le rôle est connu, ce qui ne représente que 1.5% de notre génome. La fonction des 98.5% paires de bases restantes est inconnue.
Les méthodes de séquençage, beaucoup plus rapides aujourd’hui, ont permis de séquencer des génomes d’individus provenant du monde entier. La comparaison de ces génomes montre une différence de l’ordre de 3 millions de nucléotides sur les 3 milliards de nucléotides du génome, soit une différence d’environ 0.1%. Deux humains de n’importe quelle région du globe ont donc un génome identique à 99.9%.
A titre de comparaison, il y a plus de diversité génétique entre 2 chimpanzés d’un même clan qu’entre deux humains de 2 continents différents.
Comparer nos génomes : étude de la diversité du génome humain
Bien que globalement très similaires, nos génomes diffèrent d’un individu à l’autre. Ces différences génétiques entre humains, issues de mutations, sont en majorité des différences ponctuelles, c’est-à-dire des variations d’un seul nucléotide (SNP : single nucléotide polymorphism).
Certaines parties du génome sont cependant très variables d’un individu à l’autre. Ce sont des régions de l’ADN qui comportent de nombreux SNP (régions hypervariables de l’ADN). La probabilité pour que deux individus aient exactement la même séquence de nucléotides dans ces régions hypervariables est extrêmement faible. Le séquençage de ces régions permet donc (en théorie) de distinguer tous les humains actuels et est utilisée dans le système judiciaire pour identifier un individu à partir d’une petite quantité d’ADN (technique d’empreinte génétique : séquençage et comparaison de régions hypervariables de l’ADN).
exposé : Utilisation de la génétique dans le système légal : vous êtes coupable ! (d’après vos gènes).
La comparaison des SNP de deux individus permet aussi de retracer leurs liens de parenté. En effet, lorsque deux individus possèdent de nombreux SNP similaires, l’explication la plus probable est que ces SNP soient hérités d’un même ancêtre commun qui en était porteur. Le partage de nombreux SNP indique donc des relations de parentés (tests de paternité/maternité/fratries).
L’étude des SNP permet aussi de mesurer la distance génétique entre individus et de retracer ainsi leur généalogie. En effet, certaines régions du génome contiennent de nombreux SNP transmis ensemble à la descendance (haplotypes). L’étude de ces régions permettent donc de définir des groupes de parentés, ou haplogroupes (exposé Sommes nous tous descendants de Gengis Khan ?)
Enfin, pour chaque SNP (zone de l’ADN = gène), il existe 4 versions possibles (4 allèles : A, C, G, T). Le fait de posséder l’un ou l’autre de ces nucléotides peut n’avoir aucun impact sur le fonctionnement cellulaire ou être à l’origine de différents allèles fonctionnels d’un même gène.
L’étude de certains gènes montre que certains allèles sont plus fréquents dans certaines populations. Cela s’explique généralement par le fait que ces allèles apportent / ont apporté un avantage à ces populations (sélection naturelle actuelle ou passée). Ainsi, l’allèle permettant de digérer le lactose à l’âge adulte est beaucoup plus fréquent dans les populations qui ont été amenées à consommer du lait que dans les populations qui n’en consomment pas.
(exposé Tolérance au lactose ; Phénotypes sanguins et altitude : quand un caractère neutre devient avantageux ; Drépanocytose et paludisme : quand un allèle délétère devient avantageux )
vidéo : Les bourrelets de l’évolution : être gros est-il un avantage ?
Parfois, le fait que certains allèles soient plus fréquents dans certaines populations s’explique par d’autres mécanismes évolutifs, comme le nombre d’individus à l’origine des populations (effet fondateur – dérive génétique, vus en seconde)
Peuples insulaires et évolution ; vidéo : Ces peuples prouvent la théorie de l’évolution
Certains allèles et/ou certaines combinaisons de SNP peuvent donc être des marqueurs génétiques de certaines populations géographiques. Il est donc possible d’avoir une idée +/- précise des origines géographiques d’un individu en comparant ses SNP avec les SNP représentatifs de certaines populations géographiques (exposé Connaitre ses origines grâce aux tests génétiques).
Enfin, certains allèles peuvent augmenter le risque de développer certaines maladies, tels les cancers. Si séquencer ces portions du génome permet de savoir si nous sommes porteurs ou non de ces allèles (et donc de savoir que nous avons un risque plus élevé de développer un cancer), cela ne permet pas de savoir si nous le développerons ou non (exposé Utilisation de la génétique dans le système de santé : vous tomberez malade ! d’après vos gènes).
Retracer l’histoire de notre espèce : études des génomes de nos ancêtres
(exposé Comment la génétique permet de mieux comprendre l’histoire du genre Homo)
Le fait que la diversité génétique entre humains soit très faible indique une origine commune récente de toutes les populations humaines actuelles.
Il est même possible de dater l’époque à laquelle notre lignée (à l’origine de tous les humains actuels) c’est séparée de celle de nos lignées sœurs. En effet, sachant qu’a chaque génération, une cinquantaine de mutations sont transmises à la descendance, il est possible de calculer le nombre de générations qui séparent deux individus et donc de dater la séparation des lignées auxquelles ils appartiennent : c’est le principe de l’horloge moléculaire.
Afin de retracer notre histoire, deux haplotypes sont principalement étudiés : l’haplotype porté par le chromosome Y (transmis par voie paternelle masculine uniquement du père à ses fils) et l’haplotype porté par le chromosome mitochondrial (ADN propre aux mitochondries, présent dans l’ovule et transmis par voir maternelle : de la mère à tous ses enfants).
Grace au principe de l’horloge moléculaire, il est possible de dater les chromosomes ancestraux à partir de l’étude de la diversité des haplotypes actuels. Il apparait que le chromosome Y ancestral (« chromosome Y Adam ») et le chromosome mitochondrial ancestral (« Eve mitochondriale) datent d’il y a – 200 000 ans.
De plus, l’étude de la diversité allélique des actuelles populations humaines permet de retracer les migrations de nos ancêtres. En effet, lors d’une migration, les individus qui partent de la population d’origine sont peu nombreux. Ils représentent donc une faible proportion de la diversité allélique totale présente dans la population d’origine, c’est-à-dire un faible nombre d’allèles différents pour chaque gène.
Le fait que l’on observe une diminution de la diversité génétiques des populations humaines lorsque l’on s’éloigne de l’Afrique de l’Est indique que l’intégralité de l’humanité actuelle est issue de descendants ayant migré depuis une origine géographique commune située en Afrique de l’Est.
L’étude des haplotypes et les datations des fossiles permettent ainsi de retracer et de dater les grandes migrations de l’humanité (exp : Comment la génétique permet de retracer les migrations de populations ?)
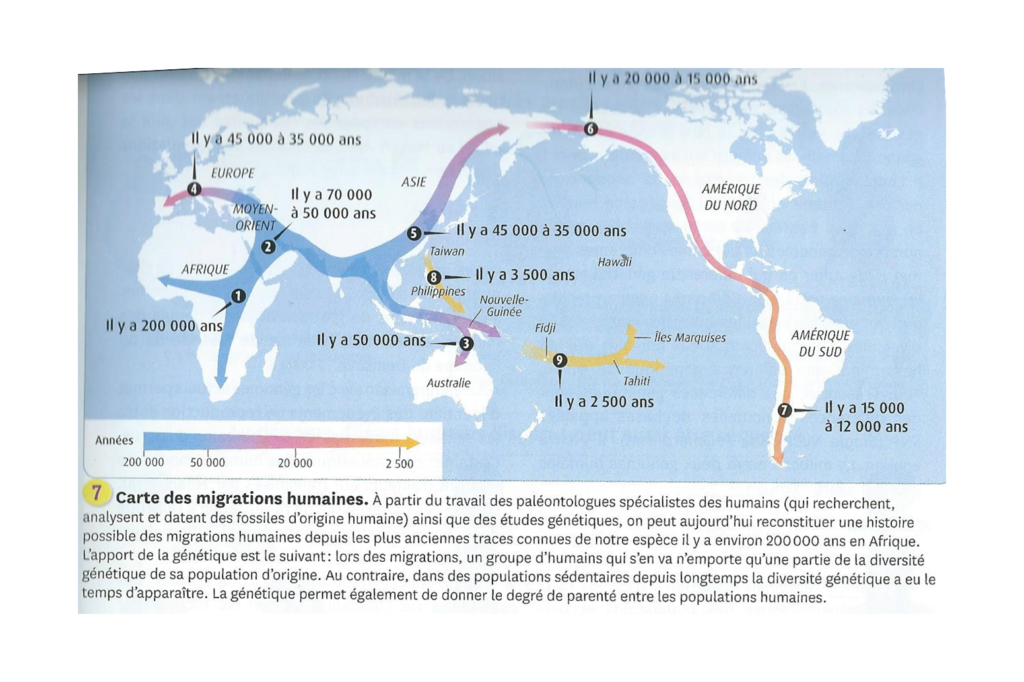
Enfin, le séquençage, l’analyse et la comparaison du génome appartenant à nos ancêtres et à des espèces aujourd’hui disparues (ADN fossile) permet de retracer les parentés entre les différentes espèces du genre Homo et de retracer les principales étapes de l’histoire de notre espèce (Homo sapiens).
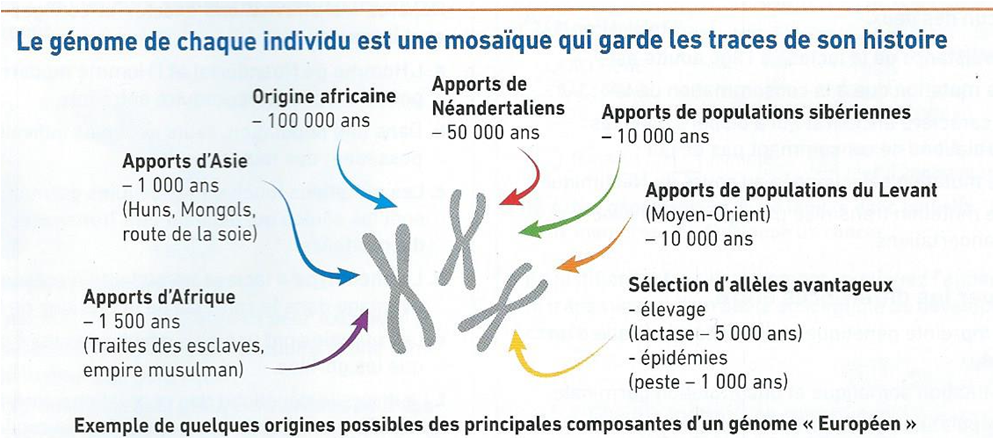
Ces études ont notamment mis en évidence que des hybridations ont eu lieu entre notre espèce (Homo sapiens) et d’autres espèces du genre Homo au cours de notre histoire évolutive.
Ainsi, le génome des populations européennes et asiatiques actuelles possède 1 à 3% d’ADN provenant d’Homo néanderthalensis (l’Homme de Néanderthal) et le génome des populations asiatiques et océaniques actuelles possède 1 à 6% d’ADN provenant d’Homo denisovensis (l’Homme de Denisova), deux espèces d’Homo actuellement disparues mais qui se sont hybridées avec nos ancêtres (exposé : Néandertal et Cro-Magnon : que c’est il passé il y a 40 000 ans ?).
Notre génome conserve donc des traces de son histoire évolutive, qu’il s’agisse de fragments d’ADN d’espèces fossiles, de preuves de métissage avec d’autres populations ou d’allèles ayant apporté des avantages à nos ancêtres.
Enfin, si l’environnement ne peut pas modifier notre génome, il apparait qu’il peut affecter les mécanismes responsables de la régulation de l’expression de nos gènes (épigénétique). Ces modifications épigénétiques étant transmissibles aux générations futures, nos génomes peuvent aussi conserver des traces des conditions de vie de nos ancêtres
(exposé : Quand nos gènes gardent des traces de la vie de nos ancêtres : exemple de la famine hollandaise).
Pour conclure, retenons que malgré la grande diversité observable entre êtres humains nos génomes sont en réalité très similaires, et témoignent de notre histoire collective. Nous sommes tous cousins, et appartenons tous à la même espèce (Homo sapiens), elle-même métissée avec d’autres espèces sœurs du genre Homo.
| Des liens avec le passé et …le futur ! |